Letter to Editor
The Daring of Biosimilars

Nuris Ledón* and Agustín Lage
Center of Molecular Immunology, Havana, Cuba
*Address for Correspondence: Nuris Ledón, Center of Molecular Immunology, Havana, Cuba, Email: [email protected]
Dates: Submitted: 06 April 2017; Approved: 28 April 2017; Published: 01 May 2017
How to cite this article: Ledón N, Lage A. The Daring of Biosimilars. Arch Biotechnol Biomed. 2017; 1: 017-020. DOI: 10.29328/journal.abb.1001002
Copyright License: © 2017 Ledón N, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The so-called “biotechnological revolution” is changing the structure of the Pharmacopoeia [1]. The space of biological products, previously limited to blood products and vaccines, grew from the introduction of first recombinant therapeutics in the 1980s until attaining a 25% by value of the pharmaceutical market. This share is expected to reach 50% in the coming years. More than 80 biotechnology drugs have entered the market in the last ten years. It is estimated that there are more than 900 biological products on development for more than 100 diseases [2].
Biopharmaceuticals have been used especially in the treatment of chronic diseases such as autoimmunities and cancer, which require long term treatments. Biologicals have an average cost 22 times higher than chemical drugs [3]. The appearance of effective but costly drugs for long treatments is contrary to the goal of gaining access to the best treatments and the goal of achieving wide population coverage. This conflict was already faced 30 years ago with synthetic drugs, and the solution was the introduction of generic drugs, at the moment when the patents of innovators expire.
In 1984, the Hatch-Waxman Act [4] legalized an abbreviated regulatory pathway for generic drugs, which it could be registered, based on clinical information of the innovative product and without the need to repeat costly clinical trials, if the producer could demonstrate that his product was chemically equal and “bioequivalent” (with pharmacokinetic data). The consequences were that prices in many cases dropped to 10% of the original price and generic prescriptions (in the United States) went from 19% in 1984 to 86% in 2013 [5].
Similar strategy for biotechnology drugs was not adopted in the twentieth century because the majority of them were protected by patents. In fact, when World Trade Organization appeared in 1995, universal patent protection for medicines, which only existed up to then in some countries, was imposed everywhere. But at the dawn of the 21st century, many biotech patents began to reach 20 years and to expire. The intellectual property barrier (first barrier) began to collapse. At the same time, the technological barrier (second barrier) began to shrink as well. More countries, especially large emerging countries with a tradition of generic production (India, China, Brazil, Korea) began to acquire capacities for biotechnological production. Cuba began producing and exporting erythropoietin, granulocytic colony stimulating factor, interferon and other recombinant drugs in the 1990s.
Then the world moved to the third barrier: The regulatory barrier. The regulatory context for biotechnology in the 1980s was initially marked by the concept that “the process defines the product”. In the framework of this doctrine no “biosimilar” was possible, because no process is identical to others in all its details and because the details of the processes are generally protected by the companies as an industrial secrets. But this concept has also been eroded by the accelerated development of analytical techniques for biological products. The sensitivity of mass spectrometry has increased more than 1000 times since the beginning of biotechnology. A complete molecular characterization is now possible. These technologies also demonstrated that biological molecules are intrinsically variable, often showing modifications of their amino acids and their chains of carbohydrates (e.g. glycosylation, desamination). In fact, in the same product, there are molecular variations from one batch to another, and from one production facility to another in the same company [6].
Then, what should be the criteria to accept that one biopharmaceutical is the same as another, and that they are interchangeable in medical practice? This is the central question of today’s regulatory controversy around biosimilars, and it is particularly intense for monoclonal antibodies which are much larger (MW 150,000) and complex molecules than the first recombinant drugs such as insulin, interferon or erythropoietin.
The first response from regulatory agencies in the United States and Europe was the requirement for randomized, comparative clinical trials between the original innovative product and the possible biosimilars with many patients. The cost of introducing a biosimilar in the market grew up to become very close to the cost of developing an innovative product. The reduction of prices did not occur on the expected magnitude, and the entry of new lower cost products was delayed. The cost of clinical trials to demonstrate biosimilarity is estimated to be 100 times higher than the cost of a bioequivalence study of a small molecule.
The first recombinant biosimilar product was approved in Europe in 2006 and in the United States only in 2015. Twenty years after the expiration of the major patents in the United States there are only four biosimilars registered. These include filgrastim, etanercept, and two monoclonals (infliximab and adalimumab) for the treatment of autoimmune diseases. In Europe, which had a policy of greater stimulus to biosimilars, there are 25 (10 different types) [7]. However, the policies have been very different from country to country. For example, the market share of erythropoietin´s biosimilars ranges from 10% in France to 67% in Germany [8]. The experience accumulated so far has not shown significant risks of toxicity or inefficiency, nor of increased immunogenicity, as a result of the change from the original product to biosimilar.
The increasing entry of costly biopharmaceuticals in the standard treatment of chronic diseases requires a national biosimilars strategy. Biosimilars are not generic products in the strict sense, because the molecular identity with the original product cannot be guaranteed, as in generic products obtained by chemical synthesis; But they are also not novel products in the strict sense, because they are supported by an enormous amount of previous science, about the type of molecule, molecular targets and mechanisms of action. The key of the strategy is in the concept of “totality of evidence” that has been enunciated in many regulatory documents but never well operationalized.
The totality of the evidence have four components:
• The knowledge about the structure of the molecule and its mechanism of action
• Safety, Pharmacokinetics and pharmacodynamics data obtained in preclinical studies.
• The safety and efficacy data, always provisional, obtained in the first clinical trials.
• And the data that can be obtained from the close monitoring on the use of the product in terms of current medical practice.
The different functions of a therapeutic monoclonal antibody must first be investigated by in vitro, receptor binding assays and in vivo assays, which are much more sensitive than clinical trials. Likewise, the characteristics that are critical in the structure/function relationship should be established, and biosimilarity should be assessed in that context. Preclinical safety studies should also be carried out.
If these tests prove to be satisfactory, PK/PD and immunogenicity tests will be performed on interchangeability assays with patients in any one of approval indication on the drugs using a design of 3 switches, with the application of Bayesian statistics. This could be enough for the approval of a biosimilar product in the indications in which the original was approved. In addition, extensive pharmacovigilance studies post registration are necessary (Figure 1). These knowledge components complement each other and determine the implicit risk assessment in the decision to allow the product to go into the medical practice.
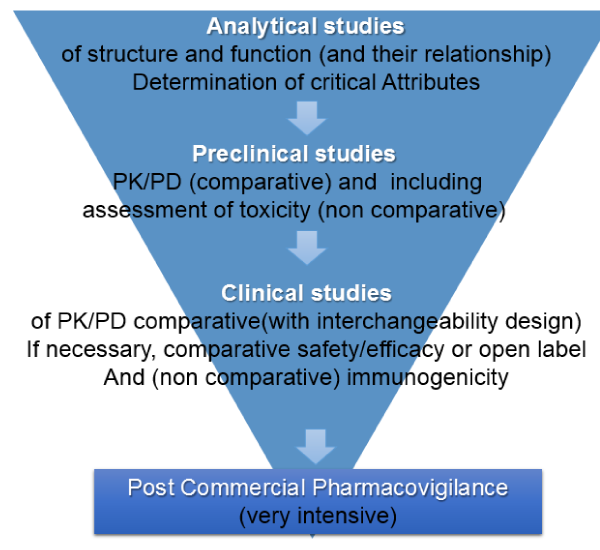
Figure 1: Proposal of Regulatory and Approval Pathway for Biosimilars.
Statistical methods for evaluating biosimilars are evolving toward greater use of Bayesian statistics [9]. Standard or “frequentist” statistical methods are designed to capture evidence from a simple study, estimating probabilities from the data in that study disregarding the previous evidence. Bayesian methods take into account the influence of previous evidence on the interpretation of data from a clinical trial, which is exactly the case for biosimilars, where in the time to start a clinical trial, there is already a large volume of evidence of the original product.
The scientific debate between the producer and the regulatory agency is a rational exercise on objective data. The subsequent step of decision-making, on the other hand, is a judgment of value on the risk/benefit balance implied in the decision to extend the use of the product, as well as in the decision not to do so. This process should include the judgment not only the producer and the regulatory agency, but also those of health system managers, government agencies, and patients or their representatives.
This value judgment cannot be independent of the context, the medical situation available and the resources available to deal with it. The “comparator” to evaluate the data on the new product has to be the real medical situation, not a hypothetical one.
The decision to introduce a new product into the medical system in a context of previous and extensive coverage with innovative products or other therapeutic products to the “state of the art” should not have the same requirements of an equivalent decision in the context of economic and organizational difficulties to access the latest treatments.
Scientific knowledge about the product to be introduced, whenever there are obtained and evaluated, is a continuous process of knowledge acquisition that never ends. The decision to contextualize pertains what is the moment, in the length of that continuum, in which we take the step of extending the use of the product to the market.
Each country, with its health system, will be take its own decision. The specific situation in Cuba is a combination of elements that do not coincide frequently: the social vocation to guarantee full coverage for all patients, the severe financial constraints together with regulatory experience and the existence of productive capacity for the development of biopharmaceuticals.
The biosimilars program must take these specificities into account.
REFERENCES
- Niosi J. Imitation and innovation new biologics, biosimilars and biobetters. Technology Analysis & Strategic Management. 2017; 29: 251-262. Ref.: https://goo.gl/1thHhm
- Evens R, Kaitin K. The evolution of biotechnology and its impact on health care. Health Aff. 2015; 34: 210-219. Ref.: https://goo.gl/m1Zfoi
- Richardson E. Biosimilars: To Encourage Competition, the Health Care Law Directs the FDA to Develop an Accelerated Approval Pathway for Follow-on Versions of Original Biologic Products. Health Aff. 2013; 1-5. Ref.: https://goo.gl/ZVhvQ7
- Kesselheim AS, Darrow JJ. Hatch-Waxman Turns 30: Do we need a Re-Designed Approach for the Modern Era? Yale J Health Policy Law Ethics. 2015; 15: 293-347. Ref.: https://goo.gl/m60BcS
- Walsh G. Biopharmaceutical Benchmarks. Nat Biotechnol. 2014; 32: 992-1000. Ref.: https://goo.gl/yCu0EM
- Schietl M, Stangler T, Torella C, Čepeljnik T, Toll H, et al. Acceptable Changes in Quality Attributes of Glycosilated Biopharmaceuticals. Nat Biotechnol. 2011; 29: 310-312. Ref.: https://goo.gl/R0vV5m
- EMA site. Available April 2017. Ref.: https://goo.gl/VAdNL
- Grabowski HG, Guha R, Salgado M. Regulatory and cost barriers are likely to limit biosimilar development and expected savings in the near future. Health Aff. 2014; 33: 1048-1057. Ref.: https://goo.gl/ZrNzbC
- Combest AJ, Wang S, Healey BT, Reitsma DJ. Alternative Statistical Strategies for Biosimilar Drug Development. Generic and Biosimilar Initiative Journal. 2014; 3: 13-20. Ref.: https://goo.gl/4Bs5ji